

INTRODUCTION
In line with Article 20 (9) of the TPD 2014/40/EU1, a guidance document outlining a reporting system for suspected adverse effects (AE) from electronic cigarettes and their refill liquids was produced by Work Package (WP) 4 entitled ‘Integration into national policies and sustainability’ of the Joint Action on Tobacco Control (JATC)2. The JATC is a European project, which lasted three years (2017–2020), with the general objective of providing support for the implementation of the TPD throughout the 27 European Union Member States (EU MS).
The reporting system described in the guidance document includes a short reporting template for the reporting of AE on human health from electronic cigarettes and refill liquids, both by Economic Operators (EO) and by National Competent Authorities (NCA), prepared by WP 7 of the JATC. The present guidance document aims at providing the necessary steps and modalities that each MS should follow if involved in the development of a system for the reporting of suspected AE on human health from electronic cigarettes and refill liquids. All the AE must be reported and notified to NCA.
The main purpose of the system for the reporting of AE is monitoring the AE from the use of electronic cigarettes (e-cigarettes) and refill liquids, in order to improve usage safety and protect the health of users by reducing the likelihood of reoccurrence of the AE. This is to be achieved by the evaluation of the reported AE and the dissemination of information, which could be used to prevent the reoccurrence or to alleviate the consequences of the AE. This guidance is intended to facilitate the uniform application and implementation of the system for reporting AE related to the e-cigarettes and/or refill liquids as previous research has noted that reporting across EU MS differs substantially3. The reporting form set out in this guidance applies to products after their placing on the market. Indeed, the availability on the web of a variety of products of uncertain quality, the continuous evolving design and changing technology of the products, strengthen the importance to detect any AE related to this practice and consequently warn users about the potential risks to health4.
AE REPORTING SYSTEM
The reporting system is based on two forms for AE reporting. This guide applies to or involves EO (manufacturer, distributor, importer), NCA, public health institutes/medical institutions (in the case they manage the surveillance system and then report to the NCA), healthcare professionals (e.g. from local healthcare units, hospitals, poison control centers, smoking cessation services, etc.) and also product users or non-users. The reporting form templates are presented in the Supplementary file.
The first form (Supplementary file Material 1) is for Economic Operators (manufacturer, importer, distributor) who receive the form from users and/or health professionals and then send the notification to the competent authority. In fact, according to Article 20 (9) of TPD, manufacturers, importers and distributors must maintain a system for collecting information about all of the suspected AE on human health of these products. The reporting to the NCA by the economic operators is mandatory.
The second form (Supplementary file Material 2) is for the users and health professionals for the voluntary reporting of AE. Users, patients and health professionals send the form directly to the competent authorities. It is recommended to report the problem also to the manufacturer/importer, or distributor shown on the label, and to the store where the product was purchased. It is not necessary to be certain of a cause/effect relationship between the AE and the use of the product(s) in question, as suspicion of an association is sufficient reason to report an event. When a manufacturer, healthcare professional, researcher, public health official, or user, reports AE incidents related to electronic cigarettes and refill liquids, most importantly contributes to public health safety and protection. The NCA’s role is to investigate the AE and take action as required; while the economic operator is responsible for reducing the harm of electronic cigarettes. It is also essential to educate consumers about modifying or adding substances (chemicals, compounds, ingredients, or combination of ingredients) other than those intended by the manufacturers and to know how these changes affect the health, frequency, and patterns of consumer use of the products. It is currently possible to purchase 0% nicotine liquids to which consumers may add nicotine (which is regulated) which therefore enables consumers to have access to large volumes of nicotine containing liquids. Electronic cigarettes can be modified by consumers as parts are widely sold, which enables consumers to modify these products5. Legislative provisions to restrict these activities would increase safety. The AE reporting system furthermore helps to identify the product or other substances including tetrahydrocannabinol (THC) and other cannabinoids6, along with other agents (i.e. diluents and other additives, nicotine, pesticides, opioids, poisons, heavy metals, and toxins)7 that may be the cause of the AE. Types of health or safety problems could include but are not limited to: fires or explosions; burns or other injuries; accidental or unintended product exposures involving children; poisonings and other toxicities; allergic reactions; any AE following use of or exposure to a product8,9.
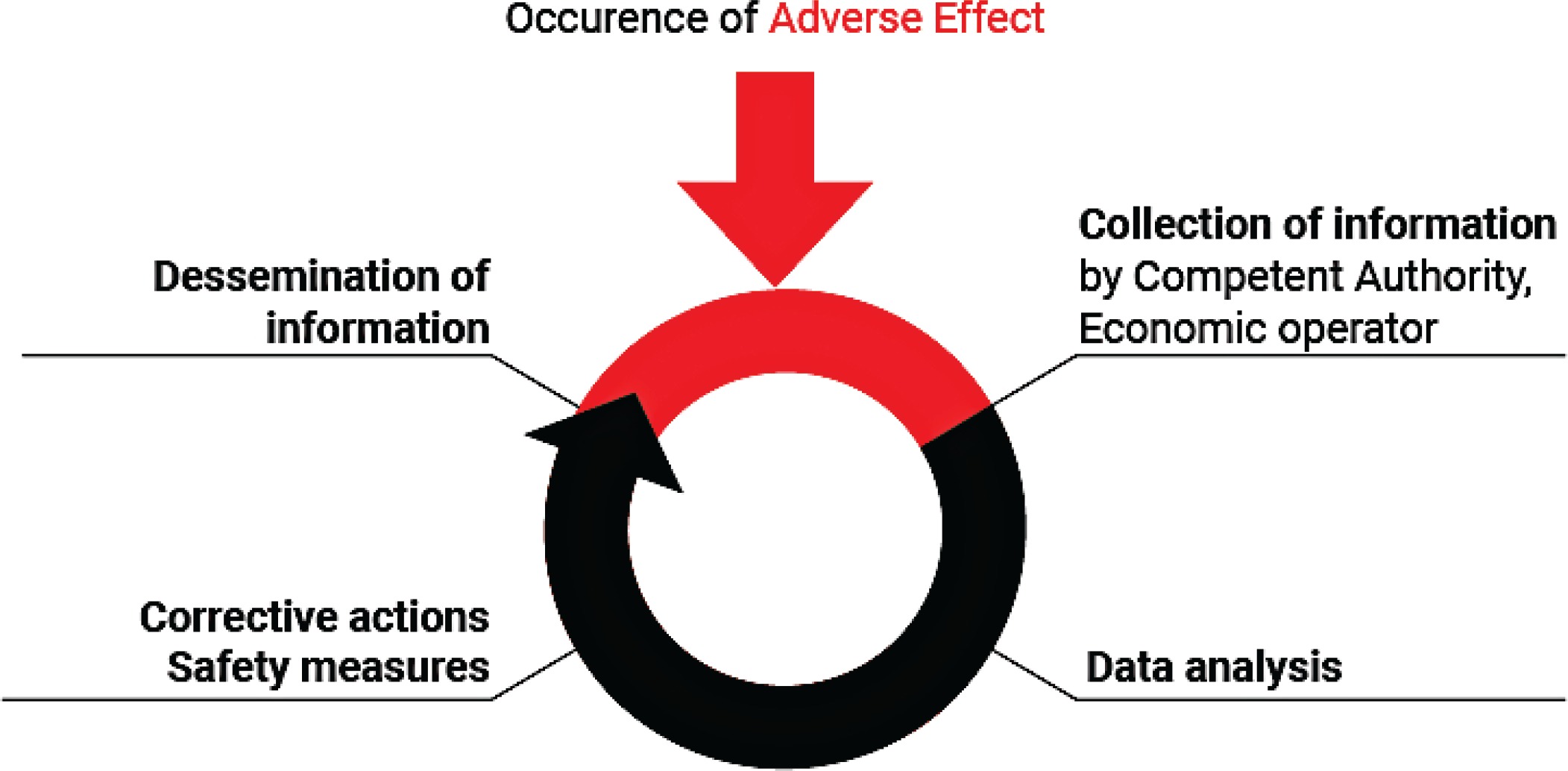
The EO receives the form from users and/or healthcare professionals and notifies the relevant NCA about AE incidents. For instance, a manufacturer may put in practice safety corrective actions as a consequence of the use of flawed or malfunctioning electronic cigarettes and/or altered refill liquid (and/or accessories). Each manufacturer submits a report to the relevant NCA for recording and evaluation. The NCA, after receiving the notification from the EO, monitors the investigation of the AE incident carried out by the manufacturer and the related corrective actions and contacts the media to notify the public (Figure 1). The NCA should then take further action that may be necessary to supplement the actions of the manufacturer. Depending on the outcome of the investigation, any information necessary for the prevention of further incidents (or the limitation of their consequences) should be disseminated by the NCA. Where an AE presents a serious risk to users from an electronic cigarette or refill liquid, a notification may need to be sent by the market surveillance authority through the Safety Gate rapid alert system, to enable the European Commission to alert other Member States10. When the notification is verified by the European Commission, it will be disseminated to market surveillance authorities via the Safety Gate as an alert for consideration and action where required. Where an NCA is not the market surveillance authority, systems should be in place to ensure that the market surveillance authorities are notified in such instances [Article 20(9) of the TPD and market surveillance legislation]1,11. EU MS should ensure that organizations and individuals involved in purchasing electronic cigarettes and refill liquids are aware that their co-operation is vital in providing the first link in the vigilance chain. In order to enhance the efficiency of the Reporting System, the NCA should encourage the reporting of AE incidents by the users and other professionals involved. Any AE incident report should be available to the other European CA and to other NCA. An overview of the process is depicted in Figure 2.
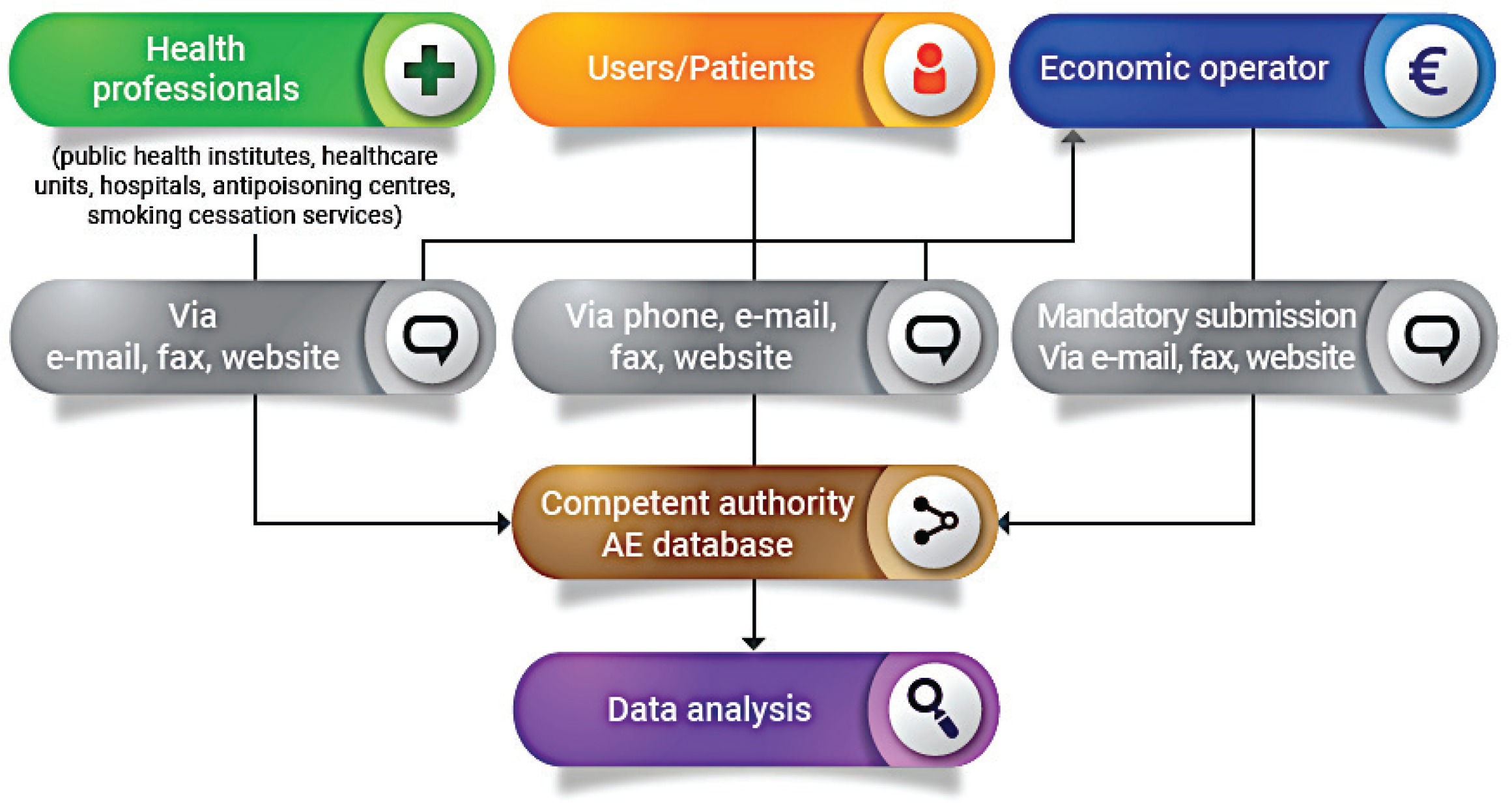
It is possible also to send the form both to economic operators and to the competent authority, and in this case it should be indicated if the form was also sent to the manufacturer/importer or distributor of the product (adding a short note in the space next to the date, in item 27 of the competent authorities form). This information helps to link the report on the AE incident to the reports filed by other sources. The AE reporting should be done even if the reporter is not sure the product caused the AE or does not have all the details, all sections that apply to the report are to be filled. The users/patients identity and privacy is held in strict confidence by NCA and protected according to the General Data Protection Regulation (EU) 2016/679 (GDPR)12.
The reporter’s identity, including the identity of a self-reporter, may be shared with the manufacturer, unless requested otherwise.
Assessment of causality in adverse effects
The association between the use of the electronic cigarette and/or refill liquid, and the occurrence of each AE shall be assessed as follows:
Unknown: the reporter has no information about the cause.
Unrelated: when the AE is not a known AE of the product; the AE has no temporal relationship with the use of the product; the AE does not follow a known response pattern to the e-cigarette (if the response pattern is previously known) and is biologically implausible; the discontinuation of e-cigarette use and reintroduction of its use does not impact on the AE; AE involves a body-site or an organ not expected to be affected by the electronic cigarette; the AE can be attributed to another cause (e.g. an underlying or concurrent illness/clinical condition, an effect of another device, drug, treatment, or other risk factors).
Unlikely: the relationship with the use of the electronic cigarette and/or refill liquid seems not relevant, and/or the AE can be reasonably explained by another cause, but additional information may be obtained.
Possible: the relationship with the use of the electronic cigarette and/or refill liquid is weak but cannot be ruled out completely. Alternative causes are also possible (e.g. an underlying or concurrent illness/clinical condition and/or an effect of another device, drug, or treatment). Cases where relatedness cannot be assessed, or no information has been obtained, should also be classified as possible.
Likely/probable: the relationship with the use of the electronic cigarette seems relevant and/or the event cannot be reasonably explained by another cause, and additional information may be obtained.
Certain: the AE is associated with the electronic cigarette and/or refill liquid use when: the AE is a known AE of the product; the AE has a temporal relationship with electronic cigarette use; the AE involves a body-site or organ expected to be affected by electronic cigarette use; the AE follows a known response pattern to the electronic cigarette use (if the response pattern is previously known); the discontinuation of electronic cigarettes and reintroduction of their use (or increase of the level of exposure) impacts on the AE; other possible causes (e.g. an underlying or concurrent illness/clinical condition and/or an effect of another device, drug, or treatment) have been adequately ruled out; and harm to the subject is due to error in usage.
Information that should be reported in the AE report
When reporting an AE, the following information should be included:
The affected person (can be a user but also a non-user);
The brand name and manufacturer/importer of the electronic cigarettes and refill liquid;
Where the electronic cigarette was purchased;
Whether the electronic cigarettes were modified in any way or whether there was a device malfunction;
Use of or exposure to tobacco products, medications, supplements, substances of abuse or toxins around the same time. Information on the use of concomitant medical products can frequently provide insight into previously unknown interactions between products, or provide an alternative explanation for the observed AE. Product names and therapies or any other medical products (drugs, medical devices, etc.) that the patient was using at the time of the AE should be listed. Products used to treat the patient after the event should not be included;
Details about the pattern of product use or exposure before the AE (duration, amount and intensity of product use) as well as the time between the latest use and the AE (this information is particularly useful in the evaluation of a suspected AE);
Dosage of nicotine, types of flavorings used or mixed substances;
Details of health effects: specific areas of the body affected, symptom progression, how long symptoms lasted, course of recovery, and any medical testing, care and treatment. In particular, a life-threatening AE should be checked if it is suspected that the patient was at substantial risk of dying at the time of the AE or if the use or continued use of the device or other medical product might have resulted in the death of the patient. Disability caused by an AE means a significant, persistent or permanent change, impairment, damage or disruption in the patient’s body function/structure, physical activities and/or quality of life;
Underlying health conditions, relevant medical history. Knowledge of other risk factors can help in the evaluation of a reported AE;
Whether product use continued or not after the AE; and
Whether the AE was reported elsewhere, such as to the manufacturer/importer or distributor (item 27 of the form, information should be added in the blank space next to the date).
Relevant medical documents, photos and other documents that include or supplement this information can also be included or reported, ensuring patient confidentiality as per Helsinki Declaration guidelines.
Reporting timelines
For voluntary reporting, users and health professionals must report to NCA or economic operator or to both, as soon as possible. In the case the economic operator gets the notification of AE by the user or health professional, it must report to the NCA as follows:
For AE that indicate an imminent risk of death, serious injury, or serious illness, which require prompt remedial action: report immediately but not later than 2 calendar days.
For any other AE: report immediately but not later than 7 calendar days following the date of awareness by the economic operator of the new reportable AE or of new information in relation to an already reported AE.
The NCA in charge of the surveillance system should issue a complete report every six months. The report should include: conclusions relating to safety and quality of electronic cigarette device and liquids, AE on health, eventual risks of self-made device and liquids, and eventual warnings about specific brands of electronic cigarettes and liquids. If the health/safety issue is serious, an urgent report should be immediately issued and information disseminated to the public. As described above, i.e. that ‘any AE incident report should be available to the other European CA and other NCA’, a network should be established in order to ensure the simultaneous communication and exchange of information among NCAs.
Statistical analysis and use of the reporting data
Statistical analysis should be performed with conclusions drawn by someone with appropriate expertise (NCA or Public Health Institution). The data and conclusions derived from the AE reporting form may result in corrective or preventive actions to reduce risk. For example, a manufacturer can change the labelling/instructions for use, the manufacturing processes, the device design; and competent authorities may take action to bring products into compliance, withdraw and recall products and use sanctions available to them to protect public health.
CONCLUSION
The ‘Reporting system of suspected adverse effects (AE) from e-cigarette and refill liquids’ has been developed to monitor the adverse effects on health among electronic cigarette users or non-users. Each MS can adjust it on country-level aspects and to make the information flow process even more convenient. If MS or stakeholders have any suggestions to improve the reporting system, they may suggest improvements to the European Commission. The national authorities may consider the contribution of a section of the EU-CEG fee or e-cigarette product taxes for the development and implementation of a reporting system on suspected AE from electronic cigarettes and refill liquids. Public reporting burden for this collection of information is estimated to average one hour per response for both voluntary and mandatory report, including completing and reviewing the collection of information. Other times (and costs) are necessary for gathering and maintaining the data received, to analyze them and evaluate the information in order to disseminate information to the public urgently or periodically. National circumstances and priorities should be taken into account during the development and implementation process.